Suivez-nous:
Processus de Qualification
ESM > Processus de Qualification
Processus de Qualification des Équipements Pharmaceutiques
Dans l’industrie pharmaceutique, la qualification des équipements est essentielle pour garantir que les équipements utilisés dans la production respectent les normes strictes de qualité, de sécurité et de conformité aux règlements. Le processus de qualification se divise en plusieurs étapes clés, de la spécification des besoins utilisateurs (URS) jusqu’à la validation finale par le Qualified Person (QP). Voici un aperçu détaillé du processus et de chaque étape impliquée :
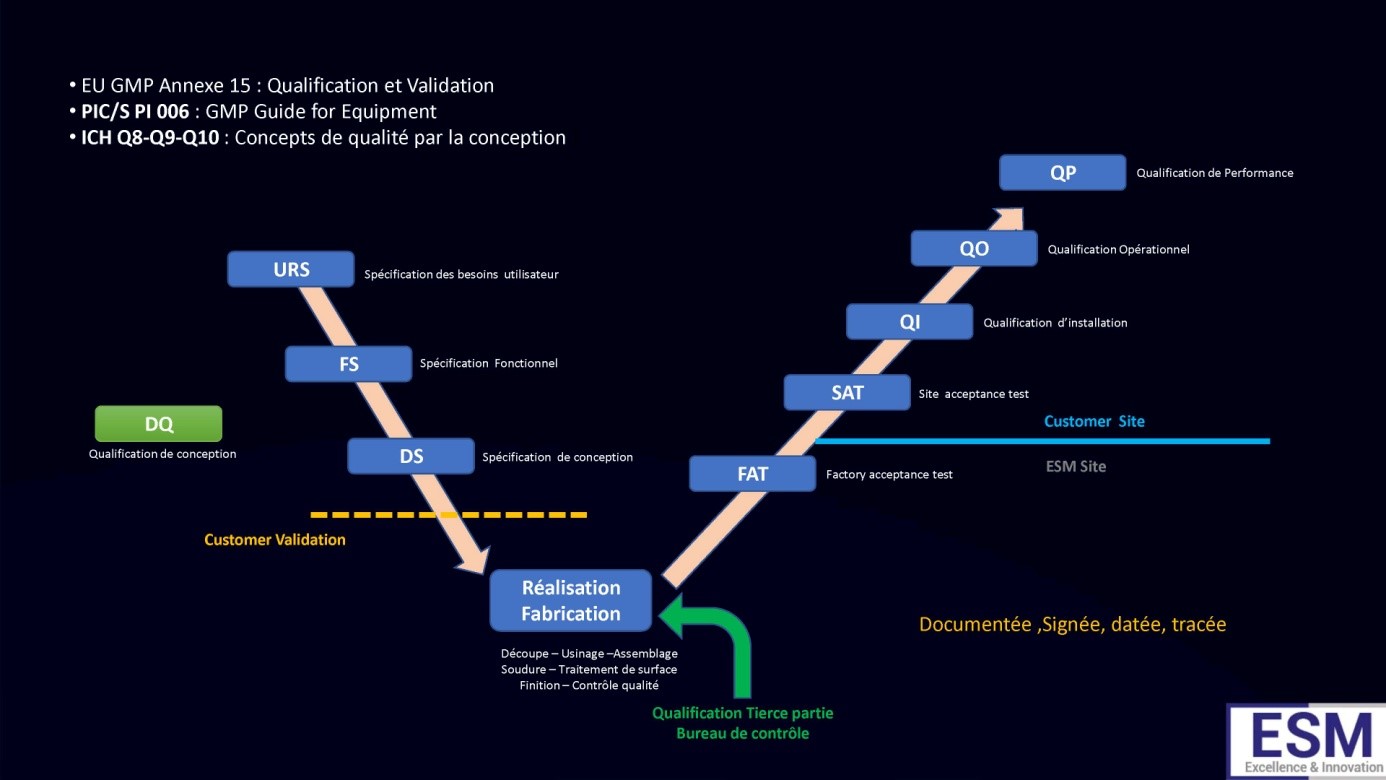
User Requirements Specification (URS) - Spécification des Besoins Utilisateurs
Objectif : Définir les exigences fonctionnelles, techniques, et réglementaires de l’équipement.
- Exigences fonctionnelles : Capacités de production, performance de l’équipement, conditions d’utilisation (température, pression, etc.).
- Exigences réglementaires : Conformité aux normes GMP, FDA, ISO, etc.
- Critères de sécurité : Systèmes de sécurité, protection de l’opérateur, prévention des risques de contamination.
- Exigences de nettoyage et de stérilisation : Processus CIP (Clean-In-Place), SIP (Sterilize-In-Place).
- Documentation : Attente de la documentation technique complète pour l’équipement.
Functional Specification (FS) - Spécification Fonctionnelle
Objectif : Détail des fonctions et caractéristiques spécifiques de l’équipement.
- Fonctions de base : Définir les tâches que l’équipement doit accomplir, telles que le stockage, le mélange, la filtration, etc.
- Définition des interfaces : Spécification de l’intégration avec d’autres équipements et systèmes, par exemple SCADA, PLC.
- Performance attendue : Critères de performance pour garantir que l’équipement sera capable de répondre aux besoins de production.
Design Specification (DS) - Spécification de Conception
Objectif : Définir les caractéristiques techniques détaillées de l’équipement basé sur l’URS et le FS.
- Matériaux : Choix des matériaux (ex. inox 304L, 316L, 904L).
- Conception mécanique : Plans détaillés des composants et de l’assemblage de l’équipement.
- Systèmes de contrôle : Spécification des systèmes de contrôle, de monitoring, et des interfaces homme-machine (HMI).
- Conformité réglementaire : Conformité avec les bonnes pratiques de fabrication (GMP), directives FDA, etc.
Réalisation de l’Équipement
Objectif : Fabrication de l’équipement selon les spécifications de conception.
- Fabrication : Fabrication selon les spécifications définies dans le DS.
- Assemblage et installation : Préparation et montage des composants, vérification des connexions.
- Tests de pré-fabrication : Vérification des performances avant livraison sur site.
Factory Acceptance Test (FAT) - Test d'Acceptation en Usine
Objectif : Vérifier la conformité de l’équipement avec les spécifications avant son expédition.
- Tests fonctionnels : Vérification des fonctions de l’équipement dans l’environnement de production simulé.
- Tests de performance : Évaluation de l’efficacité et de la capacité de l’équipement à répondre aux exigences de performance.
- Inspection : Vérification de la conformité des matériaux, des dimensions, et de la finition de l’équipement.
Site Acceptance Test (SAT) - Test d'Acceptation sur Site
Objectif : Tester l’équipement une fois installé sur le site pour confirmer qu’il fonctionne conformément aux spécifications.
- Tests d’installation : Vérification des systèmes de raccordement, alimentation, et sécurité.
- Tests d’intégration : Vérification de l’intégration de l’équipement dans le processus de production.
- Validation : S’assurer que l’équipement fonctionne dans les conditions réelles d’exploitation.
Qualification d'Installation (QI)
Objectif : Vérifier que l’équipement est installé conformément aux spécifications du fabricant et aux exigences des utilisateurs.
- Inspection visuelle : Contrôle des installations et de l’équipement pour vérifier la conformité physique.
- Vérification des utilitaires : Vérification des raccordements électriques, d’alimentation en eau, en gaz, etc.
- Documentation : Contrôle des documents d’installation et des certificats de conformité.
Qualification Opérationnelle (QO)
Objectif : Vérifier que l’équipement fonctionne comme prévu dans des conditions réelles d’utilisation.
- Tests fonctionnels : Vérification des paramètres de fonctionnement (température, pression, vitesse, etc.).
- Sécurité : Vérification des dispositifs de sécurité (alarme, verrouillage, arrêts d’urgence).
- Enregistrement des données : Collecte de données de performance pour confirmer que l’équipement répond aux spécifications.
Qualification de Performance (QP)
Objectif : Confirmer que l’équipement fonctionne selon les exigences de performance à long terme et dans des conditions de production réelles.
- Validation des performances : Vérification que l’équipement atteint les performances requises en termes de productivité, qualité et rendement.
- Tests de productivité : Analyse des rendements de production, des cycles de production, et des conditions de fonctionnement.
- Traçabilité : Validation de la conformité aux exigences de traçabilité pour garantir la conformité réglementaire.
Validation Finale par le Qualified Person (QP)
Objectif : La validation finale est effectuée par une Qualified Person (QP) pour garantir la conformité aux bonnes pratiques de fabrication et aux exigences légales.
- Vérification de la conformité GMP : La QP confirme que l’équipement respecte toutes les exigences réglementaires, y compris la sécurité des opérateurs et la qualité des produits.
Document de validation : Le QP émet un certificat de validation confirmant que l’équipement est conforme et prêt à être utilisé dans le processus de fabrication.
Le processus de qualification des équipements pharmaceutiques est un ensemble de tests et de vérifications rigoureuses, allant de la spécification des besoins utilisateurs (URS) à la validation finale par le Qualified Person (QP). Chaque étape, de la spécification à la mise en service, est conçue pour garantir que l’équipement est conforme aux exigences de performance, de sécurité et de réglementation nécessaires dans l’industrie pharmaceutique.